
找出TCGA中的配对样本并正确展示数据
数据 正确 展示 找出 样本 配对 TCGA
2023-09-14 09:16:03 时间
https://mp.weixin.qq.com/s?__biz=MzIyMzA2MTcwMg==&mid=2650733030&idx=1&sn=d0e4607d8cc6e0e93022ea6a7ac5ad28&chksm=f029aa4fc75e2359595acc1dcbf12e96b66b45480cf30a944f47d84f1615831df7d93cba81d6&cur_album_id=1336402123646631937&scene=190#rd
首先TCGA中的肿瘤样本远大于癌旁样本,甚至有的肿瘤干脆就没有癌旁。
既然有癌旁,那么就有与之对应的癌组织,把他们找出来,可以做很多事情。
开始工作,加载以前的数据,这个数据在我的教程中出现过多次
rm(list = ls())
load(file = "TCGA_exprSet_plot.Rdata")
test <- exprSet[1:10,1:10]
Image 这个数据是行是样本,行名是TCGA的barcode,列是分组信息和基因名称。
就光这个格式的数据框就可以做很多事情。
Y叔推荐的这个图有毒!
图有毒系列之2
多个基因在多亚组疾病中的展示
甚至牛逼哄哄的协同犯罪(guilt of association)注释基因功能也不在话下
单基因批量相关性分析的妙用
下面我们把他们的配对信息找出来
dplyr 这个包可以对数据框进行增删改查
tibble 这个包在这里只是方便地实现行名变列,列变行名
stringr 这个包我们只用到一个功能,就是截取字符串。
配对样本来自于同一个患者,那么TCGA id的前12个字符就是一样的,我们就是根据这个信息来提取配对样本。
library(dplyr)
library(tibble)
library(stringr)
首先提取肿瘤组织的样本,同把截取后的TCGA id作为行名,distinct用于去重,因为有的患者癌症取了多个组织。
exprSet_Tumor <- exprSet %>%
rownames_to_column(var="TCGA_id") %>%
filter(sample=="Tumor") %>%
mutate(new=str_sub(TCGA_id,1,12)) %>%
distinct(new,.keep_all = T) %>%
column_to_rownames(var = "new")
看一下获得的数据
test <- exprSet_Tumor[1:10,1:10]
Image可以和一开始数据框比较一下,我把截取后的TCGA id作为行名,为了方便下面提取子集
接下来我们再把癌旁组织信息取出来
exprSet_Normal <- exprSet %>%
rownames_to_column(var="TCGA_id") %>%
filter(sample=="Normal") %>%
mutate(new=str_sub(TCGA_id,1,12)) %>%
distinct(new,.keep_all = T) %>%
column_to_rownames(var = "new")
这时候我们需要用intersect获取癌症和癌旁共有的患者,再用这个信息分别调取对应的癌症和癌旁 共有111对配对样本,
index <- intersect(rownames(exprSet_Tumor),rownames(exprSet_Normal))
exprSet_pair <- data.frame(ID=rep(seq(1,length(index)),2),
rbind(exprSet_Tumor[index,],exprSet_Normal[index,]))
test <- exprSet_pair[,1:10]
Image
第一类,我还增加了配对信息,所以从下到下是1到111,然后又是1到111,总共222行。
下面就可以作图展示了,我们选取FOXA1来看一下, R 包就是被很多人追捧的ggpubr。
data <- exprSet_pair
library(ggpubr)
ggpaired(data, x = "sample", y = "FOXA1",
color = "black",
fill = "sample",
line.color = "gray", line.size = 0.4,
ylab = "expression of FOXA1 (VST)",
palette = "npg")+
stat_compare_means(paired = TRUE)
Image
我们看到配对样本是有两条线连接起来的,所以十分方便直观。
除此之外,我们还可以用之前介绍过的方法来作图
配对样本数据如何计算及展示?
df <- data[,c("ID","sample","FOXA1")]
class <- data$sample
pv = wilcox.test(df[,"FOXA1"] ~ class,paired = T)$p.value
library(ggplot2)
library(cowplot)
pv.lab.1 <- "Wilcoxon Signed-Rank Test"
pv.lab.2 <- paste("Cancer vs Normal p =", round(pv, 3))
lab <- paste("FOXA1\n", pv.lab.1, pv.lab.2, sep="\n")
label <- ggdraw() + draw_label(lab)
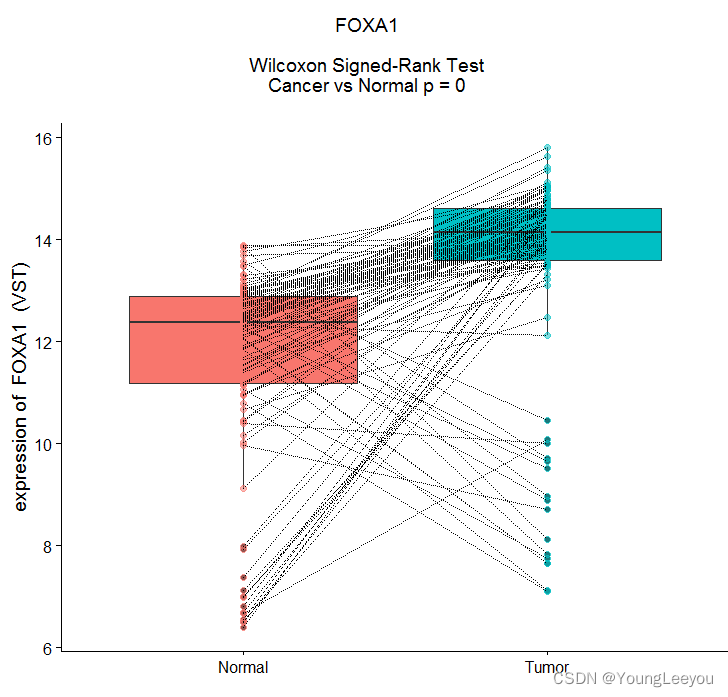
p1 <-ggplot(df, aes(sample, FOXA1,fill=sample)) +
geom_boxplot() +
geom_point(aes(colour=sample), size=2, alpha=0.5) +
geom_line(aes(group=ID), colour="black", linetype="11") +
xlab("") +
ylab(paste("expression of ","FOXA1"," (VST)"))+
theme(legend.position = "none")
plot_grid(label,p1,ncol=1, rel_heights=c(.2, 1))
Image
是得,我们可以用别人的包,也可以根据自己的喜好来定制。
现在当我们看这个数据的时候,会发现,这个连接线的方向是不一致的,上面斜着向上,还有一部分样本是斜着向下。 我们肯定会想到,这要看看亚型的信息了。
看亚型我们是有方法的,在这里。
Y叔推荐的这个图有毒!
但是,既然我们今天第一次用ggpubr,我们可以开阔一下眼界,我就现学现用
## 亚型
# Groups that we want to compare
my_comparisons <- list(
c("Basal", "Normal"),
c("Her2", "Normal"),
c("LumA", "Normal"),
c("LumB", "Normal")
)
# Create the box plot. Change colors by groups: Species
# Add jitter points and change the shape by groups
ggboxplot(
exprSet, x = "subgroup", y = "FOXA1",
color = "subgroup",
add = "jitter"
)+
stat_compare_means(comparisons = my_comparisons, method = "t.test")
Image
这里就一目了然了,确实FOXA1在不同亚型的表现不一样。
我用同一个数据,写了5个以上的帖子,实际上就是阐明一个观点,
尽管漂亮的图表是刚需,无论基金还是文章都需要,却是初学者最不应该学的。初学者应该把有限的精力用在数据清理上。R语言的特点就是R包多,我们只要把数据调整到R包需要的格式,就可以轻易做出漂亮的图片。
我们一开始常常会执着于漂亮的图片,可是最终发现,拥有属于自己的数据才是最重要的。
在这个系列里面,只要我们获取到了正确的数据,然后再用R语言把他调整成一开始的样子,所有剩下的工作就是调包,调包,调包。
稍有能力的朋友,会嘲笑初学者叫调包侠,我反倒是鼓励大家成为调包侠,但是在那之前,最应该掌握的还是数据处理的能力,这在哪里都是基本功。
我如果培训别人也是基于这个思路: