
论文解读HN-PPISP:一种基于MLP-Mixer的蛋白质-蛋白质相互作用位点预测混合网络
Title:HN-PPISP: a hybrid network based on MLP-Mixer for protein–protein interaction site prediction
期刊:Briefings in Bioinformatics
影响因子:13.994
中科院分区:Q1
出版日期:2022年11月19号
DOI:10.1093/bib/bbac480
Github:https://github.com/ylxu05/HN-PPISP
摘要
动机:蛋白质-蛋白质相互作用(PPI)位点预测的生物学实验方法对于理解生化过程的机制至关重要,但费时费力。随着深度学习(DL)技术的发展,最流行的基于卷积神经网络(CNN)的方法被提出来解决这些问题。虽然已经取得了重大进展,但这些方法在编码蛋白质序列中每个氨基酸的特征方面仍然存在局限性。现有的方法不能有效地探究位置特异性评分矩阵(PSSM)、二级结构和原始蛋白质序列的本质。在PPI站点预测中,如何有效地对PPI上下文进行建模并兼顾预测仍然是一个有待解决的问题。此外,PPI特征的长距离依赖关系也很重要,这对于许多基于CNN的方法来说是非常具有挑战性的,因为CNN的固有能力很难优于变形金刚等自回归模型。
结果:为了有效地挖掘PPI特征的性质,提出了一种新型的混合神经网络HN-PPISP,该混合神经网络集成了用于局部特征提取的多层感知器(MLP-Mixer)模块和用于全局特征捕获的两级多分支模块。该模型的优点是Transformer、TextCNN和Bi-LSTM是PPI现场预测的有力替代方案。一方面,这是先进的Transformer(即MLP-Mixer)与基于序列的PPI预测的混合网络的首次应用。另一方面,与现有方法对全局特征进行整体处理不同,本文提出的两阶段多分支混合模块首先对输入特征赋予不同的注意分数,然后通过不同的分支模块对特征进行编码。在第一阶段,对不同的改进注意模块进行杂交,分别从原始蛋白质序列、二级结构和PSSM中提取特征。在第二阶段,设计了一个多分支网络来并行地聚合来自两个分支的信息。这两个分支通过一些操作(如TextCNN、Bi-LSTM和不同的激活函数)对特征进行编码并提取依赖项。在真实世界公共数据集上的实验结果表明,我们的模型在七个显著的基线上始终实现了最先进的性能。
背景
对于蛋白质相互作用位点的超预测大致分为三类:
(1) protein–protein docking,
(2) structure-based interface prediction
(3) sequence-based interaction site determination
在我们的模型中研究了最常用的特征,如原始蛋白质序列、进化信息和二级结构。Murakami和Mizuguchi[16]基于位置特定评分矩阵(Position Specific Scoring Matrix, PSSM)和预测可达性(Predicted Accessibility, PA),创建了具有核密度估计的朴素贝叶斯分类器。
本文工作简介:
结合两级多分支网络的MLP-Mixer用于PPI站点预测。据我们所知,这是MLP-Mixer在PPI站点预测领域中第一个有效提取局部特征长依赖性的应用。
•我们提出了一个两阶段多分支模块来提取有用的全局特征。在第一阶段,用两个不同的注意力模块捕获原始蛋白质序列、二级结构和PSSM的特征。第二阶段设计多特征交叉网络,对特征进行有效编码。
•在三个真实世界的公共数据集上进行了实验,实验结果和理论分析表明,与七个显著的基线相比,我们的模型实现了最先进的性能。
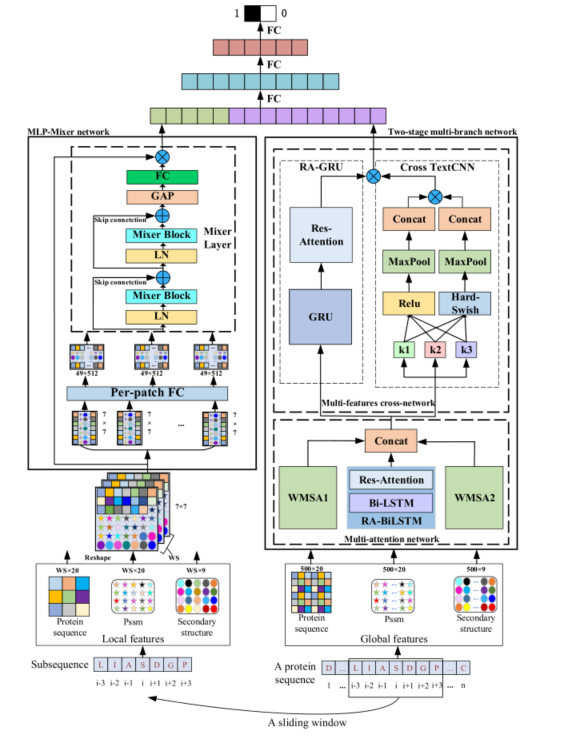
数据集与方法
数据集
我们总共使用了四个基准数据集来评估我们方法的性能。三个基准数据集Dset_186、Dset_72[16]和PDBset_164[23]均为蛋白质序列,均来自蛋白质数据库[40]。将这三个基准数据集融合为一个数据集,与[32,33]中的数据集相同,在本文中称为Dset_186_72_PDB164。另一个基准数据集是Dset_448[41]。采用与[32]相同的方法生成蛋白质原始序列对应的PSSM和DSSP,得到缩减后的数据集Dset_331,共有331个有效蛋白质数据。所以在这项工作中,我们使用了一个融合数据集,即Dset_186_72_PDB164和Dset_331数据集来进行实验。并将这两个数据集按照1:6的比例分别分为测试集和训练集。下面对这些数据集进行了更详细的描述。
数据集对应的序列长度如表一所示
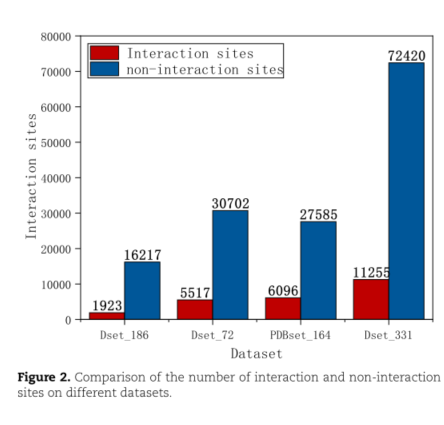
四个数据集的交互作用位点和非交互作用位点数量如图2所示。
方法
特征输入
我们利用进化信息、二级结构和原始蛋白质序列来编码蛋白质序列中每个氨基酸的特征。在生物进化过程中,蛋白质序列中的残基可能会丢失、增加或调整。在PSSM提取信息的基础上,构造特征向量来表示样本数据,并引入进化信息,表示20种氨基酸在该位置出现的概率,丰富特征向量信息。PSSM可以通过PSI-BLAST软件本地生成,也可以通过POSSUM服务器在线生成。蛋白质二级结构是通过运行蛋白质二级结构定义(DSSP)程序生成的,是指肽链的主链原子沿某一轴线盘绕或折叠而形成的特定构象,即肽链主链原子的空间排列,不涉及氨基酸残基的侧链。一个九维的单热向量被用来编码它们。前8个维度对应8种二级结构状态,分别为310-helix (G)、α-helix (H)、π-helix (I)、β-strand (E)、β-bridge (B)、β-turn (T)、bend (S)、loop or不规则(L)。最后一个维度表示没有二级结构状态信息,因为DSSP文件中可能没有某些氨基酸的二级结构状态。原始蛋白质序列,包括氨基酸及其位置信息,在实验效果[32]中起着重要作用。
MLP-Mixer网络
提出了MLP-Mixer网络来关注蛋白质的局部特征。MLP-Mixer网络的输入特征是由滑动窗口生成的子序列、子pssm和子二级结构。MLP-Mixer网络包括两个部分:Per-patch全连接(Per-patch FC)层和MLPMixer模块。每补丁FC层用于对输入特征进行嵌入式表示。首先,我们对输入子序列、子pssm和次二级结构特征进行拼接,然后将其转换为7×7×WS (WS为窗口大小)特征矩阵。然后,我们执行Per-patch FC,将每个补丁嵌入49×512维度。接下来,我们使用Mixer层进一步提取特征。一个Mixer层包括一个全连接(FC)层、全局平均池化(GAP)层、两个MLP-Mixer块和两层归一化(LNs)。MLP-Mixer块的结构如图3所示。一个MLPMixer模块包括一个高斯误差线性单元(GELU),两个FC层和两个Dropout层。MLP-Mixer块可以表示为:
结果
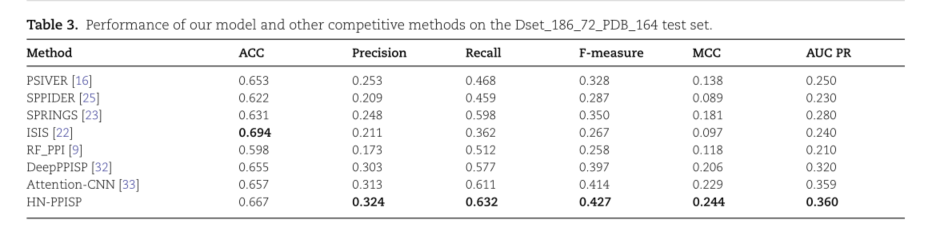
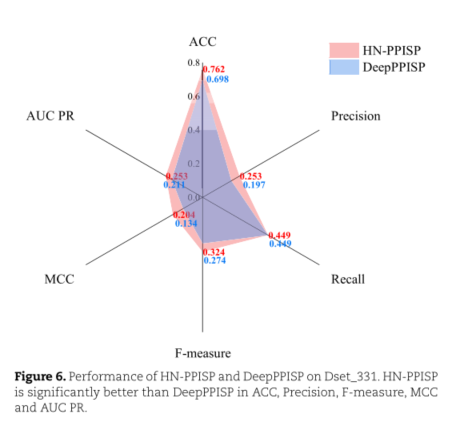
HN-PPISP和DeepPPISP在AUC上的的比较。
对于这个图以后倒是可以学一下,感觉好看又新颖。
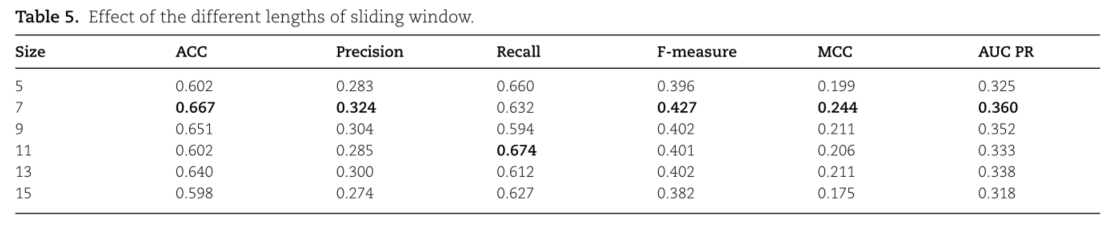
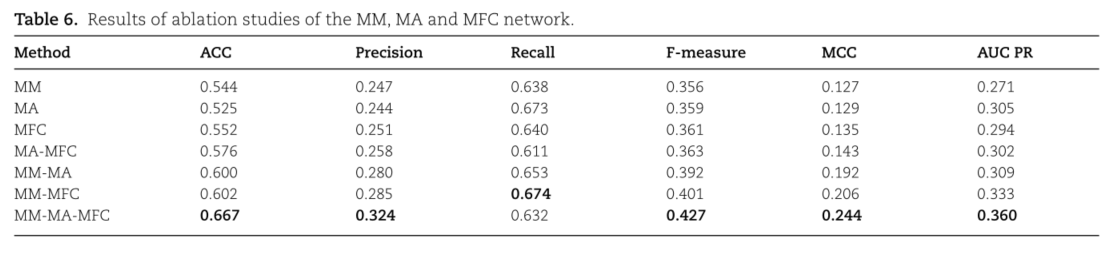
消融实验
证明滑动窗口选7最好
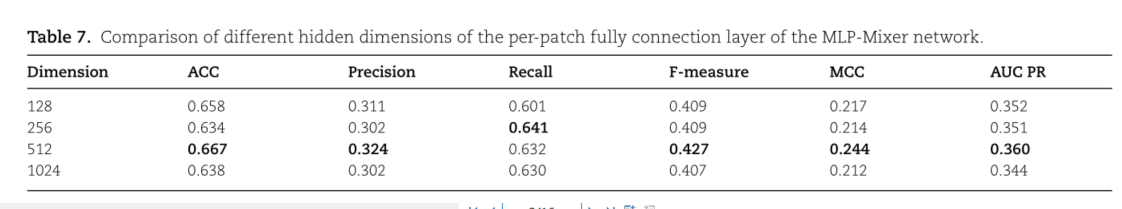
网络采用512维效果最好
加减乘除融合机制--乘法效果最好
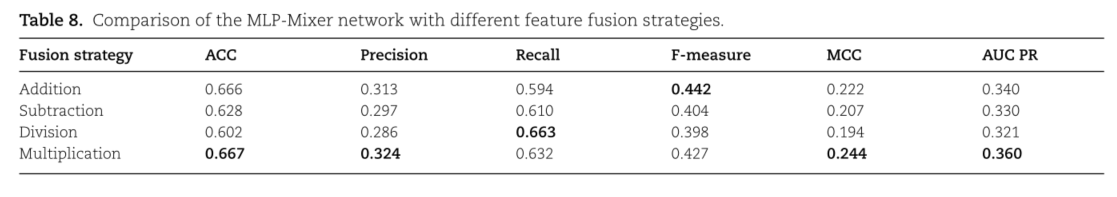
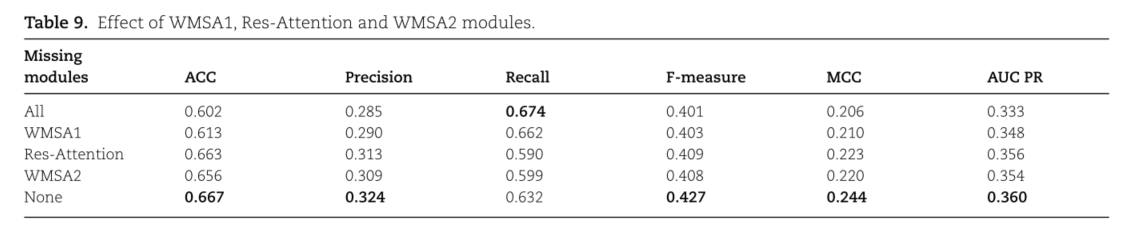
WMSA1、Res-Attention和WMSA2模块的作用
结论
相关文章
- 阿里出品的Sentinel到底是个什么玩意?
- Hadoop:从初出茅庐的小象变身行业巨人
- 亚马逊和微软协议扩大 包括BizSpark和SQL Server2012
- 微软 Windows10 任务栏搜索将深度整合 Edge 浏览器,支持原生黑暗模式
- 中小企业部署移动商业智能系统的五大要点
- 暂停升级!解决蓝屏、死机的Windows10更新翻车 用户安装失败
- 微软提前推送Windows 10 21H1更新:这些客户开始升级
- Windows10下发补丁修复打印机抽风:没想到雪上加霜
- 微软寄予厚望的Windows 10X欲推迟发布:需继续完善
- Windows10近年最重大升级近了!全新功能再次曝光
- 微软Windows10 21H1 预览版提升文件资源管理器性能:修复 explorer.exe 高内存占用
- 微软 Windows10 Dev 预览版 21337 发布:1000+ 游戏自动 HDR ,虚拟桌面大改进
- 微软确认:Windows 7/8.1免费升Windows 10依然有效
- 看Hadoop解决数据处理的三大瓶颈
- 福利仍在,微软 Windows7 免费升级Windows10 系统
- Windows 10 21H1下发新版:解决资源管理器卡内存/CPU问题
- 蓝屏后续:微软发布Windows10可选质量更新
- Windows 10下发新补丁 修复蓝屏、打印出错等问题
- 微软发布 KB500156X 累积更新,修复 Windows10打印机蓝屏 Bug
- Windows10三月更新蓝屏翻车!教你如何修复这大Bug